处理过程
表达文库分析
estep1: barcode/UMI提取
转录组文库结构:
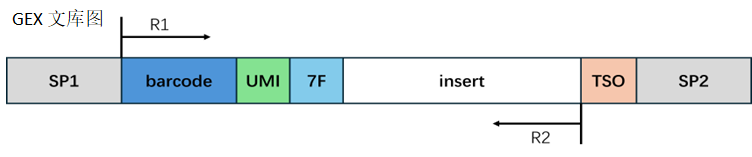
barcode 处理:
根据结构设计,取出相应序列。当取出的barcode序列在白名单中时,我们认为它是有效barcode,计入有效barcode的reads数量;当barcode不在白名单中时,我们认为它是无效barcode。
测序过程中,有一定几率发生测序错误。在提供有白名单的情况下,SeekArcTools可以尝试barcode矫正。在启用矫正时,当无效barcode一个碱基错配(一个hamming distance)的序列存在于白名单中:
只有唯一一个序列存在于白名单中:我们将这个无效barcode矫正为白名单中barcode
有多个序列存在于白名单中:我们将这个无效barcode改为read支持数量最多的序列
接头处理:
在转录组中,Read2的末端有可能会出现 7F 的反向互补序列,建库时可能引入的接头序列。我们对这些污染序列进行切除,剪切完的read2长度需要大于设定的最小长度来保证有足够的信息,准确比对到基因组的位置。如果剪切完成后的read2长度小于最小长度,我们认为这条read为无效read。
estep2: 进行比对并找到比对基因
序列比对
使用STAR比对软件将处理后的R2比对到参考基因组上。
使用QualiMap软件和转录本注释文件GTF,统计reads比对外显子、内含子和基因间区等的比例。
使用featureCounts将比对上的read注释到基因上,可以选择不同的注释规则,如链方向性和定量的feature。当使用外显子定量时,当read超过50%碱基比对到外显子区域时,认为该read来源于此外显子以及外显子对应的基因;当使用转录本定量时,当read超过50%碱基比对到转录本区域时,认为该read来源于此转录本以及转录本对应的基因。
经过上述处理后,有如下数据指标:
Reads Mapped to Genome: 比对到参考基因组上的reads占所有reads的比例(包括只有一个比对位置和多个比对位置的reads)
Reads Mapped Confidently to Genome: 在参考基因组上只有一个比对位置的reads占所有reads的比例
Reads Mapped to Intergenic Regions:比对到基因间隔区reads占所有reads的比例
Reads Mapped to Intronic Regions:比对到内含子reads占所有reads的比例
Reads Mapped to Exonic Regions:比对到外显子reads占所有reads的比例
estep3: 定量
UMI定量:
SeekArcTools以barcode为单位提取featureCounts输出的bam数据,统计注释到基因的UMI和UMI对应的reads数:
过滤掉对应UMI为单个重复碱基的reads, 例如UMI为TTTTTTTT
当read注释到多个基因,在有唯一外显子的注释时,为有效read,其他情形下都过滤掉
UMI矫正:
测序过程中,UMI也有一定概几率出现测序错误。SeekArcTools默认使用UMI-tools的adjacency方法对UMI进行矫正。
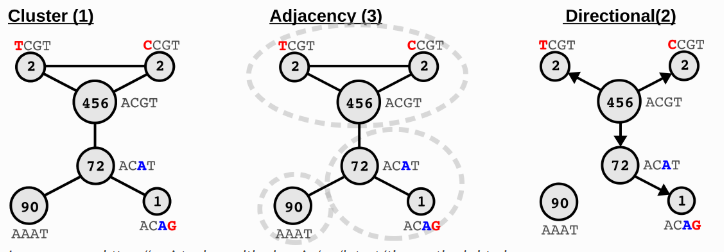
Image source: https://umi-tools.readthedocs.io/en/latest/the_methods.html
ATAC 文库分析
astep1: barcode 提取
ATAC 文库结构:
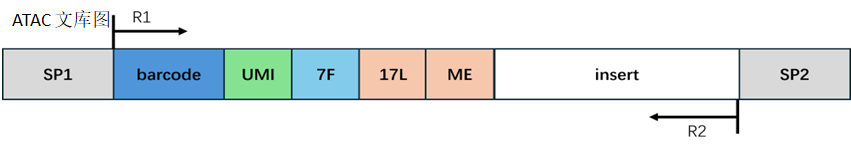
barcode 处理:
同estep1的处理方式一样。
接头和 reads 处理:
在ATAC 文库中,Read1 带有固定序列,末端有可能会出现接头序列;Read2的末端有可能会出现 固定序列的反向互补序列和接头序列。我们对这些污染序列进行切除,剪切完的read1或read2长度需要大于设定的最小长度来保证有足够的信息,准确比对到基因组的位置。如果剪切完成后的read1或read2长度小于最小长度,我们认为这条read为无效read;如果剪切完成后的read1或read2长度大于50,则截取read1或read2序列的前50bp;如果剪切完成后的read1或read2长度小于50且大于最小长度,则保留read1或read2的全部序列。
astep2: 进行比对
序列比对:
使用 bwa mem 将处理后的read1和read2比对到参考基因组上。
astep3: 定量
使用 SnapATAC2 v2.8.0 处理 astep2 输出的 bam 文件。
读取 bam ,获取ATAC 的 Sequencing 和 Mapping 相关指标。
由 bam 生成原始 fragments 文件。过滤掉线粒体相关的 fragments 后得到过滤后的 fragments 文件。
由过滤后的 fragments 文件生成 Anndata 对象。
绘制 TSS 得分分布图。
使用 MACS3 进行 call peak,并过滤重复的 peaks,输出到 peaks.bed。
根据 peaks 生成 raw_peaks_bc_matrix。
统计每个 barcode 的相关指标,输出到 per_barcode_metrics.csv。
绘制插入片段分布图。
细胞判定
生成 filtered_feature_bc_matrix 和 filtered_peaks_bc_matrix。
统计ATAC 的 Cells 和 Targeting 相关指标。
细胞判定
若指定 --min_atac_count 和 --min_gex_count:
则判定 barcode 中 UMI 数量大于 min_gex_count 值且 events 数量大于 min_atac_count 值的barcode为细胞。
若不指定:
首先,保留 barcode 中 UMI 数量大于1 或 events in peaks 数量大于 1的 barcode。
然后,过滤 "the fraction of fragments overlapping called peaks" 小于 "the fraction of genome in peaks" 的 barcode。
去除重复值。将具有相同 UMI 值和 events in peaks 值的 barcode 过滤。
将过滤后的 barcode 的UMI 值和 events in peaks 值进行 log10 转换。
根据转换后的UMI 值和 events in peaks 值,使用 KMeans 算法将 barcode 分为两群其中均值高的群为细胞,另一群则不是细胞。
将去重过滤掉的 barcode 通过判定的 barcode 映射为细胞或非细胞。
astep4: 后续分析
经过上述步骤,得到细胞的表达矩阵和ATAC 矩阵后,我们可以进行下一步的分析。
使用 Signac v1.14.0 将细胞的表达矩阵和ATAC 矩阵合并为一个 seurat object,过滤不在数据库中的 feature,分别对表达矩阵和ATAC 矩阵进行归一化、寻找高变基因、降维聚类之后寻找差异基因,进行 WNN 分群,最后计算 feature link。